

At Shiru, we are on a mission to identify proteins from plants that are functionally equivalent to animal-based food ingredients or synthetic ingredients.
This task necessitates tackling the decades-old challenge of protein function prediction, linking protein structure and function in a manner that until now has been out of reach for the food science community. Shiru is developing the means to find proteins that are functionally similar to some of the most widely used and environmentally taxing protein ingredients that exist, even if their sequences are not similar — for instance, plant proteins that have functions similar to mammalian proteins, even though they are not homologous. To this end, traditional methods based on sequence similarity are not expected to be as effective as deep learning methods. Importantly, machine learning enables us to do this at scale – cataloging food relevant functionality of millions of proteins rapidly. Previous methods to find compelling plant or non-animal protein alternatives have relied on “trial and error” methods that catalog at most dozens of proteins via difficult to generate empirical data.
Shiru is using novel machine learning approaches to better perform two tasks:
- Finding similar proteins to a target protein
- Predicting a protein function
Both tasks require accurate representations of proteins. At Shiru, we “represent” proteins by describing them as numerical vectors that capture the maximum amount of information about a protein. One’s representation could be expert-defined (usually the result of a combination of meaningful features computed by bioinformatic pipelines) or learned (usually by letting statistical models compress protein data into a single vector — e.g. encoder/decoder architectures). For the machine learning team at Shiru, we are mainly interested in the latter.
Why representations
The act of learning representations does not require the data to be labeled. Therefore, the research community leverages “big-data” to learn accurate generic representations that can be used for varying tasks. Those representations are relatively easy to work with and allow the use of a plethora of different models when it comes to a specific task (e.g. predicting a protein’s function).
Representation models pretrained on a vast amount of data can be used in two ways:
- Direct use of the representations they produce as input features.
- Fine tuned for a more specific use case.
Since the representation model already went through the effort of learning the low level protein features, using pre-trained models saves researchers a great amount of training time and doesn’t require as much data. This is known as transfer learning.
Previous work
A very successful method for learning protein representations is to train a language model (e.g. Transformer (Vaswani et al.) on millions of protein sequences (Rives et al.). This is often done using BERT (Rives et al.; Devlin et al.), a technique that trains the Transformer to fill the missing residues in a protein sequence. By doing this, the Transformer is forced to learn a representation that captures implicitly important ‘concepts’ of protein language. Those representations can be seen as an abstraction from which the protein sequence is projected. For example, two proteins with different sequences will have similar embeddings if the language model finds similarities in their previously-learned ‘concepts’.
For a while now, the proteomics community has tried to leverage evolutionary information to perform better at prediction tasks or learning representations. The valid prediction is that the invariants in protein sequences across different species give information, on which are located the most important residues, as well as an important structural signal. For this reason, many models (eg. MSA-Transformer) now are designed to handle Multiple Sequence Alignments (MSA). They work in a similar fashion to sequence-only models, but have been shown to better capture the structural information (Rives et al.; Devlin et al.; Rao et al.).
Representations from structure
We saw that models were able to learn useful representations from sequence and MSA; however, sequences and MSAs are only a proxy of what proteins truly are: 3-dimensional molecules that participate in complex interactions. If we see a protein as a graph, where each node is a residue and each edge the relationship between those residues (e.g. distance, AA-AA relationship), we withhold significantly more information.
So far, the proteomics community hasn’t had a lot of incentive to learn protein representations from graphs due to the lack of structural data (only 181,000 published structures as opposed to hundreds of millions of available sequences). However, DeepMind recently announced a partnership with EMBL to predict the structure of 135 million proteins. It is clear that this tremendous amount of data will allow us and others to develop models that are better at predicting protein functions and learning representations. At Shiru we are leveraging AlphaFold 2 predicted structures to learn more accurate protein representations. Below, we show graph information that was extracted from a protein using AlphaFold 2 predictions.
So far, the proteomics community hasn’t had a lot of incentive to learn protein representations from graphs due to the lack of structural data (only 181,000 published structures as opposed to hundreds of millions of available sequences). However, DeepMind recently announced a partnership with EMBL to predict the structure of 135 million proteins. It is clear that this tremendous amount of data will allow us and others to develop models that are better at predicting protein functions and learning representations. At Shiru we are leveraging AlphaFold 2 predicted structures to learn more accurate protein representations. Below, we show graph information that was extracted from a protein using AlphaFold 2 predictions.
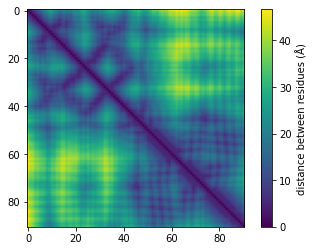
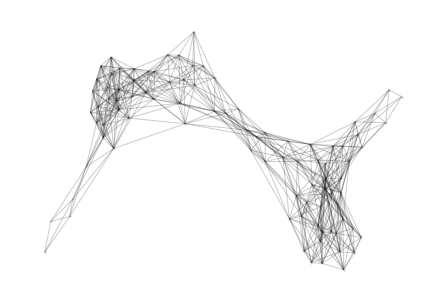

Once the graphs are computed for many sequences, we apply graph-embedding techniques that learn better representations. We are aiming to show that those new representations have significantly higher function-predicting power.
Function prediction is the primary focus going forward in multiple academic research and biotech applications. Here at Shiru, we are developing deep-learning methods that leverage the recently introduced protein structure prediction methods to better predict a protein’s food-related function. How a protein functions inside the host organism is typically distinct from its role in a food product (e.g. acting as an emulsifier, binder, texturizer, etc). These critical tools are what will enable Shiru to deliver the most functional, high performance ingredients to the food industry that ultimately result in the best foods without consumer, animal, or environmental sacrifice.
Click here to learn more about our ground-breaking work leveraging AlphaFold.
References
- Devlin, Jacob, et al. “BERT: Pre-Training of Deep Bidirectional Transformers for Language Understanding.” Proceedings of the 2019 Conference of the North, Association for Computational Linguistics, 2019, doi:10.18653/v1/n19-1423.
- Rao, Roshan, et al. “MSA Transformer.” bioRxiv, bioRxiv, 13 Feb. 2021, doi:10.1101/2021.02.12.430858.
- Rives, Alexander, et al. “Biological Structure and Function Emerge from Scaling Unsupervised Learning to 250 Million Protein Sequences.” bioRxiv, bioRxiv, 29 Apr. 2019, doi:10.1101/622803.
- Vaswani, Ashish, et al. Attention Is All You Need. 12 June 2017, http://arxiv.org/abs/1706.03762.


